Project: Structural Protein Sequences Classification
7 minute read
![]()
![]() Status: final, Type: Project
Status: final, Type: Project
Jiayu Li, sp21-599-357, Edit
- Code:
Abstract
The goal of this project is to predict the family of a protein based on the amino acid sequence of the protein. The structure and function of a protein are determined by the amino acid sequence that composes it. In the protein structure data set, each protein is classified according to its function. Categories include: HYDROLASE, OXYGEN TRANSPORT, VIRUS, SIGNALING PROTEIN, etc. dozens of kinds. In this project, we will use nucleic acid sequences to predict the type of protein.
Although there are already protein search engines such as BLAST[^1] that can directly query the known protein families. But for unknown proteins, it is still important to use deep learning algorithms to predict their functions.
Protein classification is a simpler problem than protein structure prediction[^7]. The latter requires the complete spatial structure of the protein, and the required deep learning model is extremely complex.
Contents
Keywords: Protein Sequences, Deep learning
1. Introduction
The structure and function of a protein are determined by the amino acid sequence that composes it. The amino acid sequence can be regarded as a language composed of 4 different characters. In recent years, due to the development of deep learning, the ability of deep neural networks to process natural language has reached or even surpassed humans in some areas. In this project, we tried to treat the amino acid sequence as a language and use the existing deep learning model to analyze it to achieve the purpose of inferring its function.
The data sets used in the project come from Research Collaboratory for Structural Bioinformatics (RCSB) and Protein Data Bank (PDB)1.
The data set contains approximately 400,000 amino acid sequences and has been labeled. The label is the family to which the protein belongs. The protein family includes HYDROLASE, HYDROLASE/HYDROLASE INHIBITOR, IMMUNE SYSTEM, LYASE, OXIDOREDUCTASE, etc. Therefore this problem can be regarded as a classification problem. The input of the model is a sequence, the length of the sequence is uncertain, and the output of the model is one of several categories. By comparing DNN, CNN, LSTM and other common models, we have achieved effective prediction of protein energy supply.
2. Dataset
PDB is a data set dedicated to the three-dimensional structure of proteins and nucleic acids. It has a very long history, dating back to 1971. In 2003, PDB developed into an international organization wwPDB. Other members of wwPDB, including PDBe (Europe), RCSB (United States), and PDBj (Japan) also provide PDB with a center for data accumulation, processing and release. Although PDB data is submitted by scientists from all over the world, each piece of data submitted will be reviewed and annotated by wwPDB staff, and whether the data is reasonable or not. The PDB and the software it provides are now free and open to the public. In the past few decades, the number of PDB structures has grown at an exponential rate.
Structural biologists around the world use methods such as X-ray crystallography, NMR spectroscopy, and cryo-electron microscopy to determine the position of each atom relative to each other in the molecule. Then they will submit this structural information, wwPDB will annotate it and publish it to the database publicly.
PDB supports searching for ribosomes, oncogenes, drug targets, and even the structure of the entire virus. However, the number of structures archived in the PDB is huge, and finding the information may be a difficult task.
The information in the PDB data set mainly includes: protein/nucleic acid source, protein/nucleic acid molecule composition, atomic coordinates, experimental methods used to determine the structure. Structural Protein Sequences Dataset: https://www.kaggle.com/shahir/protein-data-set/code
Protein dataset classification: https://www.kaggle.com/rafay12/anti-freeze-protein-classification
RCSB PDB: https://www.rcsb.org/
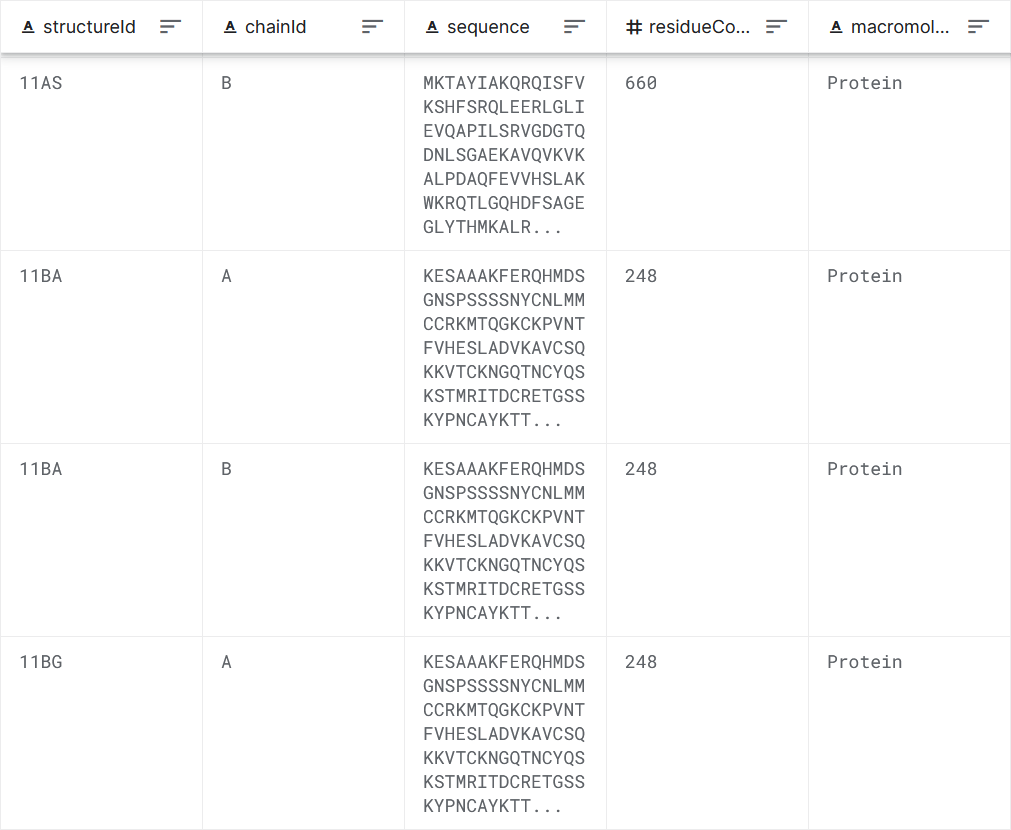
Figure 1: Data set sample.
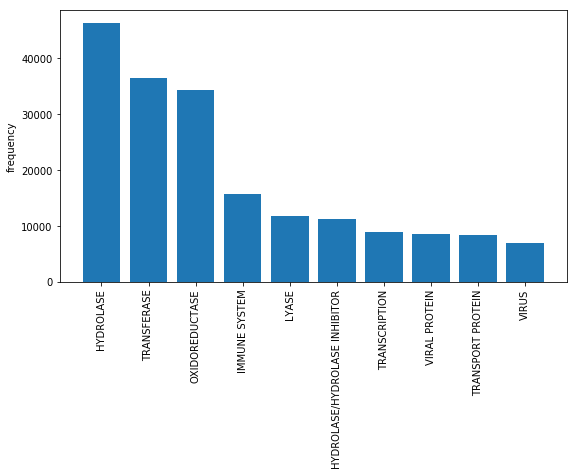
Figure 2: The frequency of appearance of different labels.
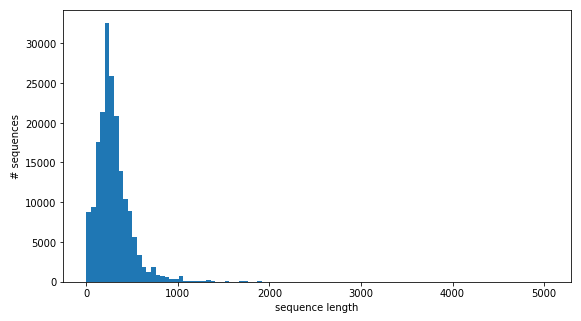
Figure 3: The length distribution of amino acid sequences.
3. Deep learning algorithm
Two deep learning models, CNN and LSTM2, are mainly used in this project. In addition, the Word Embedding algorithm is also used to preprocess the data.
Among these models, the CNN model comes from the Kaggle website3 and will be used as a test benchmark. We will try to build a simpler but more accurate model with an accuracy rate of at least not lower than the test benchmark.
3.1 Word Embedding
A word embedding is a class of approaches for representing words and documents using a dense vector representation.
Iin an embedding, words are represented by dense vectors where a vector represents the projection of the word into a continuous vector space.
The position of a word within the vector space is learned from text and is based on the words that surround the word when it is used.
The position of a word in the learned vector space is referred to as its embedding.
Two popular examples of methods of learning word embeddings from text include: Word2Vec4 and GloVe5.
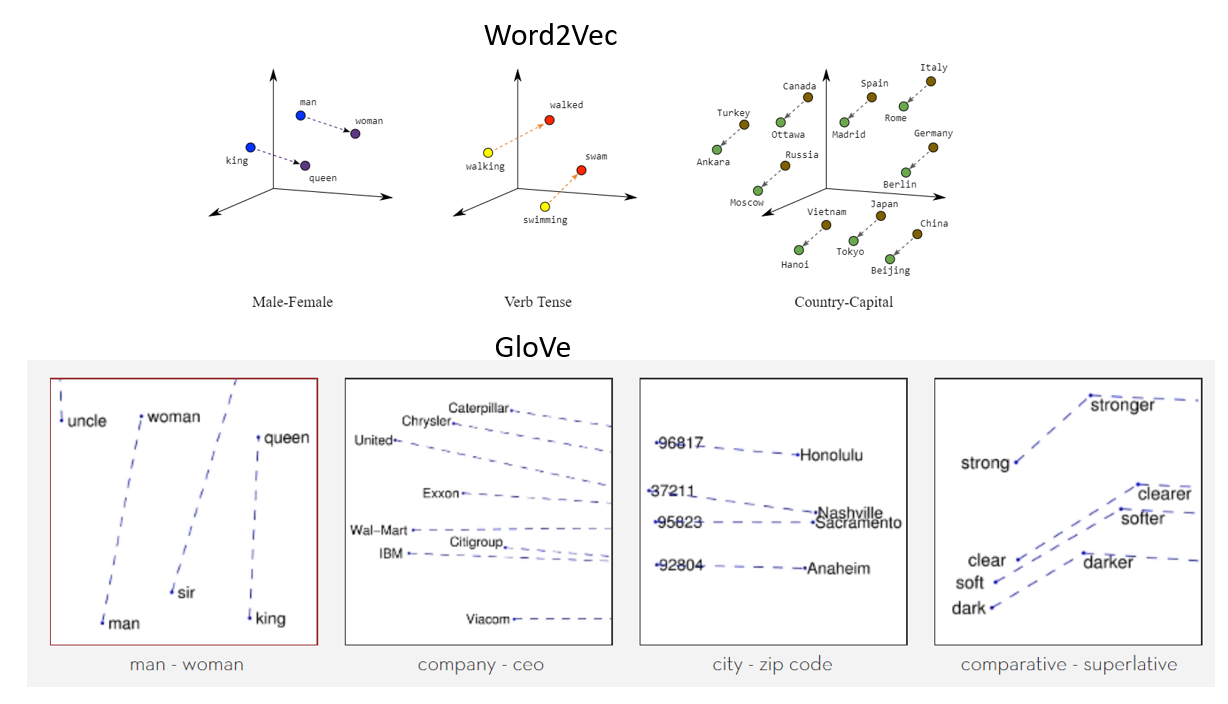
Figure 4: Word2Vec and GloVe.
3.2 LSTM
Recurrent Neural Network (RNN) is a neural network used to process sequence data. Compared with the general neural network, it can process the data of sequence changes. For example, the meaning of a word will have different meanings because of the different content mentioned above, and RNN can solve this kind of problem well.
Long short-term memory (LSTM) is a special kind of RNN, mainly to solve the problem of gradient disappearance and gradient explosion during long sequence training. Compared to ordinary RNN, LSTM can perform better in longer sequences.
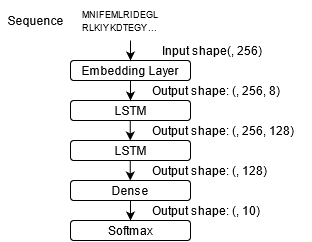
Figure 5: LSTM.
4. Benchmark
4.1 Compare with test benchmark
3 is a highly accurate model on the Kaggle website. It is currently one of the models with the highest accuracy on this data set. In this project, CNN model in 3 will be used as a test benchmark. We use categorical cross entropy as loss function.
| Model | CNN | LSTM |
|---|---|---|
| #parameters | 273,082 | 203,226 |
| Accuracy | 91.6% | 91.9% |
| Training time | 7ms/step | 58ms/step |
| Batch size | 128 | 256 |
| Loss | 0.4051 | 0.3292 |
Both the CNN model and the LSTM model use the word embedding layer for data dimensionality reduction. After testing, the result is that LSTM uses fewer parameters to achieve the same or even slightly higher accuracy than the benchmark. However, due to the relatively complex structure of LSTM, its training speed and guessing speed are slower.
In order to make a fair comparison with the test benchmark, we only selected the 10 most frequent samples as the data set, which is also the original author’s choice. Therefore, the accuracy of more than 90% here is only of relative significance, and does not mean that the same accuracy can be achieved in practical applications (the data set usually has more categories).
4.2 The impact of the number of labels on accuracy
In order to further test the performance of LSTM on different data sets, we further increased the number of labels, gradually increasing from 10 labels to 20 labels. Figure 6 shows the effect of the number of labels on accuracy.
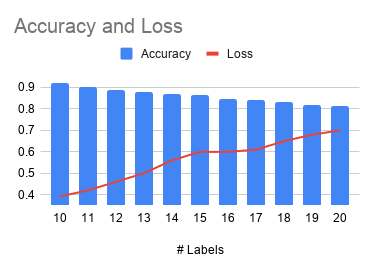
Figure 6: The impact of the number of labels on accuracy.
Note that due to the limitation of the data set, the number of samples belonging to different labels is different. If we want to balance different categories, we have to shrink the data set, which will affect the accuracy. This is one of the limitations of this test.
5. Conclusion
The deep learning model gives a prediction accuracy of more than 90% for the 10 most common protein types. If the number of label is increased to 20, the accuracy rate will drop to 80%. The traditional machine learning model is difficult to deal with string data of different lengths.
6. Acknowledgments
The author would like to thank Dr. Gregor von Laszewski for his invaluable feedback on this paper, and Dr. Geoffrey Fox for sharing his expertise in Big Data applications throughout this course.
7. References
-
Sussman, Joel L., et al. “Protein Data Bank (PDB): database of three-dimensional structural information of biological macromolecules.” Acta Crystallographica Section D: Biological Crystallography 54.6 (1998): 1078-1084. ↩︎
-
Sundermeyer, Martin, Ralf Schlüter, and Hermann Ney. “LSTM neural networks for language modeling.” Thirteenth annual conference of the international speech communication association. 2012. ↩︎
-
Kaggle, Protein Sequence Classification. https://www.kaggle.com/helmehelmuto/cnn-keras-and-innvestigate ↩︎
-
Church, Kenneth Ward. “Word2Vec.” Natural Language Engineering 23.1 (2017): 155-162. ↩︎
-
Pennington, Jeffrey, Richard Socher, and Christopher D. Manning. “Glove: Global vectors for word representation.” Proceedings of the 2014 conference on empirical methods in natural language processing (EMNLP). 2014. ↩︎